
Introduction
Drug discovery can take 10-15 years and cost billions of dollars, with high failure rates at every stage. Artificial intelligence seeks to reduce these timelines and costs by rapidly predicting molecular structures and interactions.
Over the last six or so years, numerous models have emerged in biomolecular structure prediction. Boltz-2 represents a significant advancement in this field and stands out for a number of reasons. Developed by MIT researchers as an open-source AI model, it demonstrates notable effectiveness in predicting binding affinity and comes with weights, inference, and training code released under a permissive open license. Building on the foundation of AlphaFold-3 and Boltz-1, Boltz-2 extends beyond protein structure prediction alone to handle a much broader range of molecular systems critical for drug design.
Unlike earlier tools focused primarily on proteins, Boltz-2 can predict the 3D structures of:
- Proteins and protein complexes
- Protein-ligand interactions (for understanding how drugs bind to their targets aka Binding Affinity)
- Nucleic acids (DNA/RNA)
- Small molecules and their interactions with biological macromolecules
Key Takeaways
- MIT’s Boltz-2 is fully open-source with weights, inference, and training code under a permissive license.
- Boltz-2 excels at predicting how strongly drugs bind to their targets, which is critical for therapeutic effectiveness.
- The model extends beyond predecessors AlphaFold-3 and Boltz-1 to predict structures for proteins, protein-ligand interactions, nucleic acids (DNA/RNA), and small molecule interactions.
- Boltz-2 approaches gold-standard Free-Energy Perturbation (FEP) accuracy while being far more computationally efficient.
- The training methodology involves the use of distillation from high-confidence AlphaFold-2 and Boltz-1 predictions, enhanced by Boltz-steering, which is an inference-time technique integrating physics-based potentials for improved accuracy.
Binding Affinity
Any advancement in predicting binding affinity is huge. Binding affinity refers to the strength with which small molecules bind to proteins. This is a key factor in determining a drug’s ability to engage its target and deliver a therapeutic effect. Currently, atomistic simulations such as free-energy perturbation (FEP) offer the highest accuracy for measuring binding affinity. However, their computational demands and cost make them impractical for large-scale use. While faster alternatives like docking do exist, they lack the precision needed for reliable predictions. As of now, no AI model has achieved the accuracy of FEP methods or laboratory assays in predicting binding affinity.
Data
Boltz-2, a model trained on diverse biomolecular data, improves upon Boltz-1 by incorporating ensembles from experimental and computational techniques. The training dataset includes structures from the Protein Data Bank and molecular dynamics trajectories, aiming to expose the model to local fluctuations and global ensembles. Additionally, distillation techniques are employed to augment the training data and supervision signal, utilizing high-confidence predictions from AlphaFold2 and Boltz-1.
Architecture
Boltz-2’s architecture improves upon Boltz-1 and Boltz-1x by enhancing controllability and the affinity module. It introduces method, template, and contact/pocket conditioning for more precise predictions. The affinity module predicts binding likelihood and affinity values using a PairFormer model, leveraging protein-ligand and intra-ligand interactions.
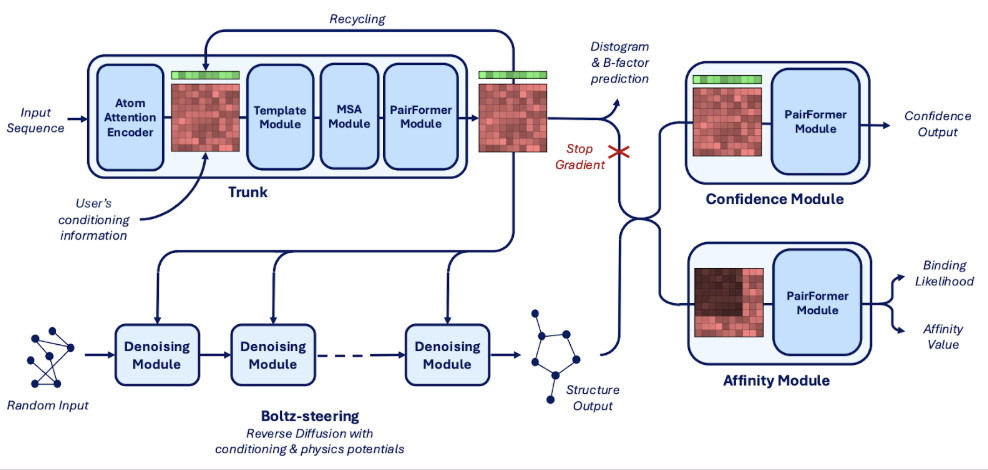
How was Boltz-2 trained?
Boltz-2’s training is divided into three phases: structure, confidence, and affinity training. Affinity training incorporates pre-computation, custom sampling strategies, and robust loss functions to improve generalization and scalability. Boltz-2 is also used to train a molecular generator, SynFlowNet, to produce small molecules with high binding scores. Alphafold-2 was distilled to expand the training set by using its high-confidence predictions on single-chain monomers.
Performance
Boltz-2, outperforms its predecessor, Boltz-1 in crystal structure prediction, particularly for RNA chains and DNA-protein complexes. It also shows competitive performance against other models like Chai-1, ProteinX, and AlphaFold3, especially in antibody-antigen structure prediction and the Polaris-ASAP challenge. Additionally, Boltz-2 demonstrates better ability to capture local protein dynamics and approaches the accuracy of free energy simulations on public benchmarks for affinity predictions.
Boltz-2 outperforms existing methods on the CASP16 affinity challenge and internal assays from Recursion. It demonstrates strong performance in virtual screening, achieving high average precision and enrichment factors on the MF-PCBA dataset. Boltz-2’s scalability and accuracy make it a promising tool for large-scale virtual screening in drug discovery.
Boltz-Steering
Boltz-steering (as part of the Boltz-1x release) is an inference-time method that incorporates physics-based potentials (also known as interatomic potentials (IPs) or empirical force fields,), which improves physical plausibility without sacrificing accuracy. This approach was also integrated within Boltz-2 to obtain Boltz-2x.
Implementation
Step 0: Power up a Droplet
Boltz-2 is designed to run on a GPU by default, though you can switch to a CPU using the --accelerator option. However, be aware that running inference without a GPU is significantly slower.
Begin by setting up a DigitalOcean GPU Droplet.
Step 1: Clone the repository
Copy and paste these commands into your terminal one by one. These commands download Boltz-2 and install the necessary dependencies.
git clone https://github.com/jwohlwend/boltz.git
cd boltz; pip install -e .[cuda]
Step 2: Prepare Your Input File
Boltz-2 needs to know what you want it to predict. You tell it this using a YAML file (a simple text file). Create a file named my_protein.yaml. Inside, you will list the sequences of the molecules you are studying.
If you aren’t sure how to format this, look at the examples/ folder inside the boltz directory.
By default, the input_path should point to a YAML file, or a directory of YAML files for batched processing, describing the biomolecules you want to model and the properties you want to predict (e.g. affinity).
For other command-line options, consult the documentation.
boltz predict input_path --use_msa_server
After running the model, the generated outputs are organized into the output directory following the structure below:
out_dir/
├── lightning_logs/ # Logs generated during training or evaluation
├── predictions/ # Contains the model's predictions
├── [input_file1]/
├── [input_file1]_model_0.cif # The predicted structure in CIF format, with the inclusion of per token pLDDT scores
├── confidence_[input_file1]_model_0.json # The confidence scores (confidence_score, ptm, iptm, ligand_iptm, protein_iptm, complex_plddt, complex_iplddt, chains_ptm, pair_chains_iptm)
├── affinity_[input_file1].json # The affinity scores (affinity_pred_value, affinity_probability_binary, affinity_pred_value1, affinity_probability_binary1, affinity_pred_value2, affinity_probability_binary2)
├── pae_[input_file1]_model_0.npz # The predicted PAE score for every pair of tokens
├── pde_[input_file1]_model_0.npz # The predicted PDE score for every pair of tokens
├── plddt_[input_file1]_model_0.npz # The predicted pLDDT score for every token
...
└── [input_file1]_model_[diffusion_samples-1].cif # The predicted structure in CIF format
...
└── [input_file2]/
...
└── processed/ # Processed data used during execution
FAQ
What is Boltz-2?
Boltz-2 is an open-source AI model developed by MIT researchers that excels at predicting the 3D structures of various biomolecular systems and, critically, predicting binding affinity—the strength with which a small molecule (like a drug) binds to a protein target.
How is Boltz-2 different from predecessors like AlphaFold-3 or Boltz-1?
Boltz-2 extends beyond protein structure prediction to handle a much broader range of systems, including protein-ligand interactions, nucleic acids (DNA/RNA), and small molecule interactions. It also incorporates sophisticated training and inference techniques (like Boltz-steering) to achieve near gold-standard accuracy in binding affinity prediction while maintaining high computational efficiency.
Is Boltz-2 truly open-source?
Yes. Boltz-2 is fully open-source, including the model weights, inference code, and training code, all released under a permissive open license.
What is “Binding Affinity” and why is its prediction important?
Binding affinity refers to the strength of the molecular interaction between a drug candidate (small molecule) and its biological target (protein). Predicting this accurately is a key factor in determining a drug’s therapeutic effectiveness and guiding lead optimization in drug discovery.
What is FEP accuracy, and how does Boltz-2 compare to it?
FEP (Free-Energy Perturbation) is a class of atomistic simulation methods considered the “gold standard” for calculating binding affinity due to its high accuracy. FEP is computationally demanding and slow. Boltz-2 approaches the accuracy of FEP methods but is far more computationally efficient, making it practical for large-scale virtual screening.
What is “Boltz-steering”?
Boltz-steering is an inference-time technique integrated into Boltz-2 (and Boltz-1x) that incorporates physics-based potentials (empirical force fields). This technique helps to improve the physical plausibility of the predicted structures and interactions without sacrificing accuracy.
What are the hardware requirements to run Boltz-2?
Boltz-2 is designed to run on a GPU for optimal performance. While running inference on a CPU is possible using the --accelerator option, it is significantly slower.
Where can I find the source code and documentation?
The repository is available on GitHub. Consult the documentation in the repository for detailed command-line options and usage instructions.
Final Thoughts
We’re excited about the drug discovery space. Boltz-2, an open-source release by MIT researchers, allows anyone to experiment with biomolecular simulation. By achieving near-gold-standard Free-Energy Perturbation (FEP) accuracy in binding affinity prediction - a critical bottleneck in drug development, mind you, while offering computational efficiency, Boltz-2 is pushing the frontier of what’s possible.
Thanks for learning with the DigitalOcean Community. Check out our offerings for compute, storage, networking, and managed databases.
About the author
Melani is a Technical Writer at DigitalOcean based in Toronto. She has experience in teaching, data quality, consulting, and writing. Melani graduated with a BSc and Master’s from Queen's University.
Still looking for an answer?
This textbox defaults to using Markdown to format your answer.
You can type !ref in this text area to quickly search our full set of tutorials, documentation & marketplace offerings and insert the link!
 This work is licensed under a Creative Commons Attribution-NonCommercial- ShareAlike 4.0 International License.
This work is licensed under a Creative Commons Attribution-NonCommercial- ShareAlike 4.0 International License.
Become a contributor for community
Get paid to write technical tutorials and select a tech-focused charity to receive a matching donation.

DigitalOcean Documentation
Full documentation for every DigitalOcean product.

Resources for startups and AI-native businesses
The Wave has everything you need to know about building a business, from raising funding to marketing your product.
The developer cloud
Scale up as you grow — whether you're running one virtual machine or ten thousand.

Start building today
From GPU-powered inference and Kubernetes to managed databases and storage, get everything you need to build, scale, and deploy intelligent applications.
